


























主な研究テーマ
- 低酸素応答による生体内エネルギー代謝制御機構の解明
- 硫黄代謝物による生体内代謝制御機構の解明
- 生理活性脂質の産生/分解機構や生体内での機能の解明
- 代謝による細胞老化の制御機構の解明
- 癌細胞特異的な代謝機構の同定と治療標的の探索
- 質量分析計を用いたプロテオーム解析・メタボローム解析(リピドーム解析も含む)による細胞内生理的/病理的イベントの分子メカニズム解明
キーワード:低酸素、代謝、硫化水素、脂質メディエーター、慢性炎症、がん、細胞老化、DNA修復、細胞周期
- 低酸素応答による生体内エネルギー代謝制御機構の解明
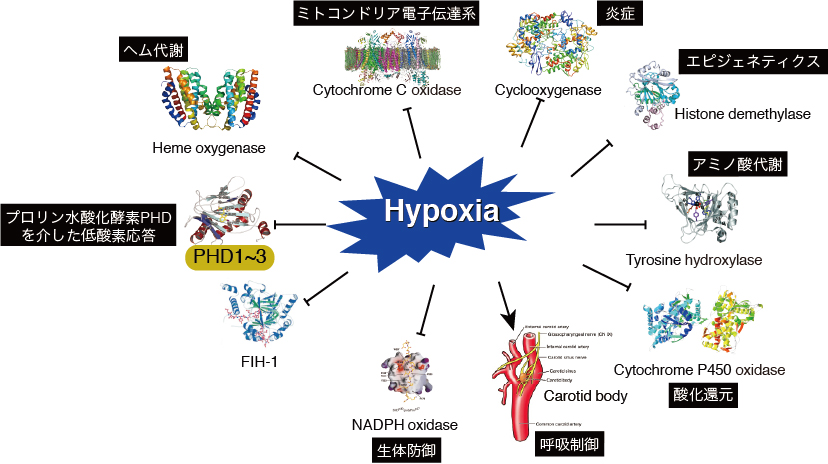
2019年のノーベル生理学・医学賞が低酸素応答の研究者3名に贈られたことからもわかるように、「低酸素の生物学」はその重要性と面白さが広く認知された研究分野です。

昨年のノーベル生理学・医学賞が低酸素応答の研究者3名に贈られたことからもわかるように、「低酸素の生物学」はその重要性と面白さが広く認知された研究分野です。
我々の身体の細胞には、利用できる酸素が限られた低酸素環境においても生存していけるような低酸素応答機構がプログラムされています。
低酸素応答は、主に転写因子HIFによって制御されていますが、そのHIFもまたプロリン水酸化酵素PHD1〜PHD3によって負に制御されているため、PHDは酸素濃度センサーとして低酸素応答をONにするのかOFFにするのかのスイッチ役を担っていると言えます。
我々のラボでは、その酸素濃度センサーPHDの遺伝子改変マウスや開発中のPHD阻害剤を用いて、低酸素応答を in vivo レベルで自由にON/OFFさせる実験モデルを構築し、低酸素応答による生体内の様々な生理的・病理イベントの制御メカニズムの解明を目指します(南嶋)。
以下に詳細を解説します。
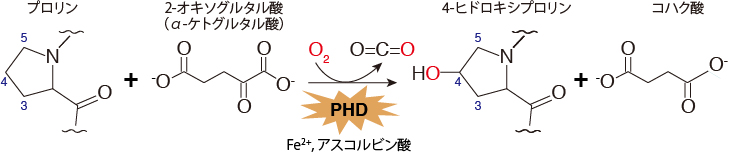
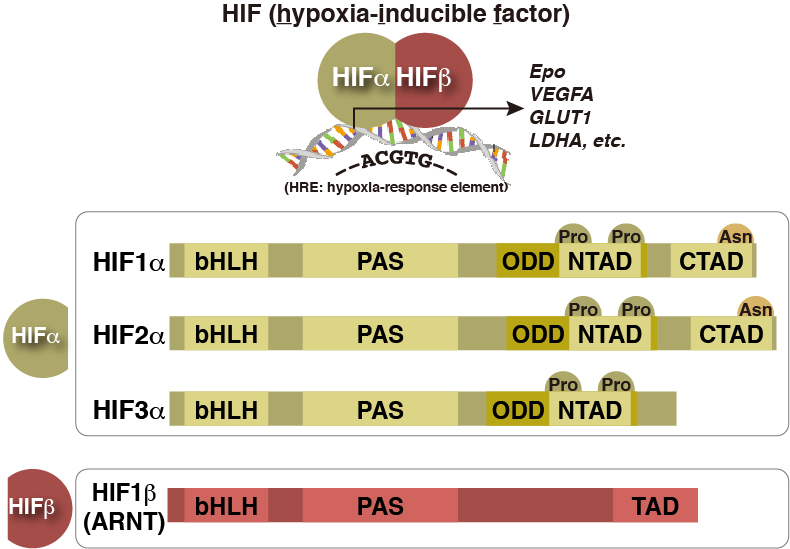
【図3】そのプロリン水酸化酵素PHDの最も有名な基質の1つがHIF (hypoxia-inducible factor)と呼ばれる転写因子のαサブユニット(HIFα)です。HIFはαサブユニットとβサブユニット(HIF1β/ARNT)がヘテロダイマーを形成することで転写因子として機能します。HIFαには3つの遺伝子が同定されており、特にHIF1αとHIF2αについてよく研究されています。HIF3αはC'末端側の転写活性ドメイン(CTAD)を欠いていることもあり、HIF1αやHIF2αと比較して転写活性が弱いようです。そのため、HIF3αはHIF1αやHIF2αの機能を抑制する役割があるとされています。実際に、低酸素応答が活性化すると、HIF3αやそのスプライシングバリアントであるIPAS(HIFに対してドミナントネガティヴに働く)の転写が誘導され、HIFの機能を抑制するような、ある種のネガティヴ・フィードバック機構が働くようです。我々の身体にはHIFの活性化しっぱなしを抑えるブレーキ機構があると言うことです。
また、低酸素=HIFではありませんし、HIF=HIF1αではありません。HIF1αだけに注目して、HIF2αやHIF3αなど一切この世に存在しないかのような表現を使う研究者を散見します(その書き方でquality journalに投稿すると、この時点で即刻rejectされます)。ユビキタスに発現しているHIF1αに対し、HIF2αは骨格筋・心筋・腎・肝・血管内皮・各種癌細胞・・・などなど、発現する細胞・組織が限られていますが、だからこそユニークな働きをしています。赤血球を増やす造血ホルモンであるEPO(エリスロポエチン)などはHIF1αではなくHIF2αによって転写されますし、VHL欠損腎細胞細胞をはじめ、HIF2αは様々な癌細胞で癌の進展に寄与しています。このように、科学的な理由なしに3つあるHIFα遺伝子のうちHIF1αにしか注目しないと言う研究はもはや科学ですらありません。
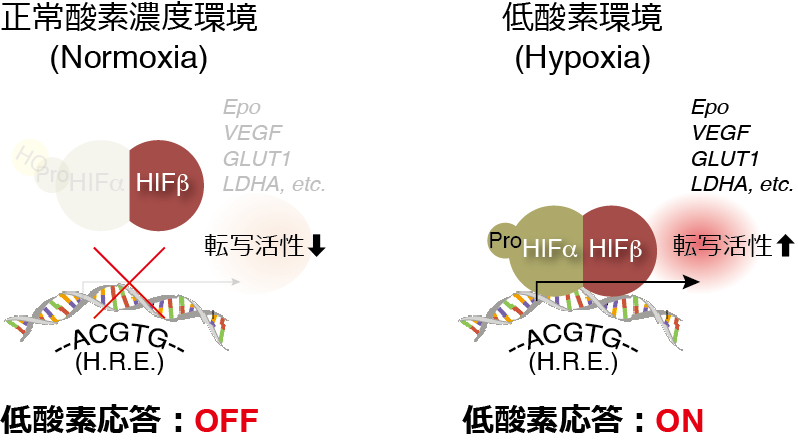
【図4】このHIFαですが、正常酸素濃度環境(normoxia)では、蛋白発現量が低く抑えられており、その結果低酸素応答はOFFとなっています。一方で、低酸素環境(hypoxia)では急激にHIFαの蛋白発現量が上昇し、低酸素応答がONになります。すなわち、低酸素応答はHIFαの発現量で制御されていることになります。
それでは、そのHIFαの発現量はどのように制御されているのでしょうか?
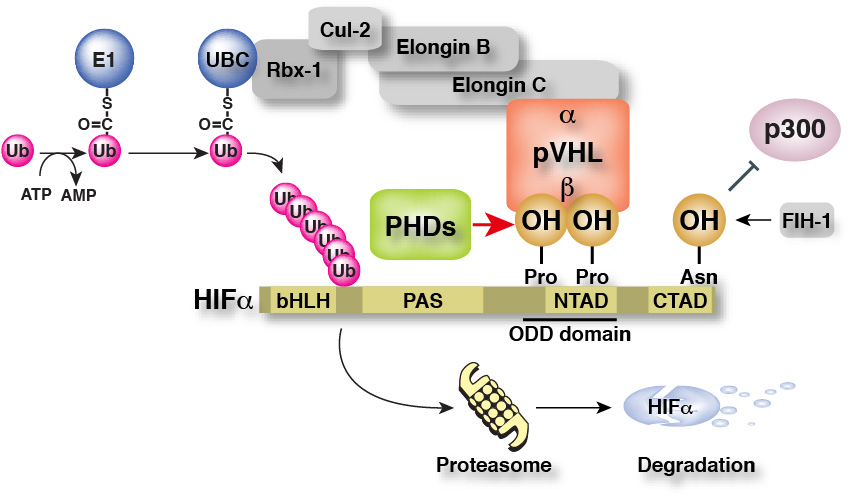
【図5A】HIFαは、N'末端側の活性化ドメインNTAD近傍の2つの特定のプロリン残基のどちらか或いは両方が水酸化されていると、pVHL(von Hippel-Lidau病の原因遺伝子VHLから転写翻訳される蛋白)のβドメインに認識され、pVHLを含むE3ユビキチンリガーゼ複合体(VBC complex)によってユビキチン化されてプロテアソームにおける蛋白分解へと導かれます。
※C末端側のアスパラギン残基も水酸化酵素FIH-1によって酸素依存的に水酸化されており、このアスパラギン残基が水酸化されると、HAT活性を持ち転写のco-activatorであるp300がHIFαに結合出来ず、転写活性が抑制されることが解っています。
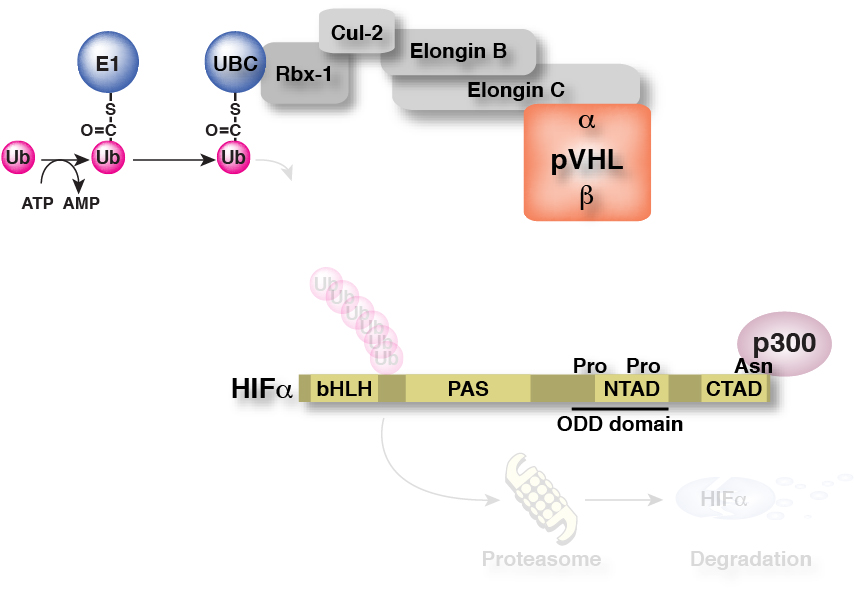
【図5B】一方で、そのHIFαの特定のプロリン残基が水酸化されていないと、pVHLはHIFαを認識して結合することが出来ないため、結果としてHIFαはユビキチン化→蛋白分解を免れて細胞内で急速に発現量が上昇します。
すなわち、低酸素応答はHIFαの発現量で制御されており、そのHIFαの発現量は特定のプロリン残基の水酸化状態に依存している、ということになります。
そして、このHIFαのプロリン残基の水酸化を制御しているのが、先ほどお話ししたプロリン水酸化酵素PHDなのです。
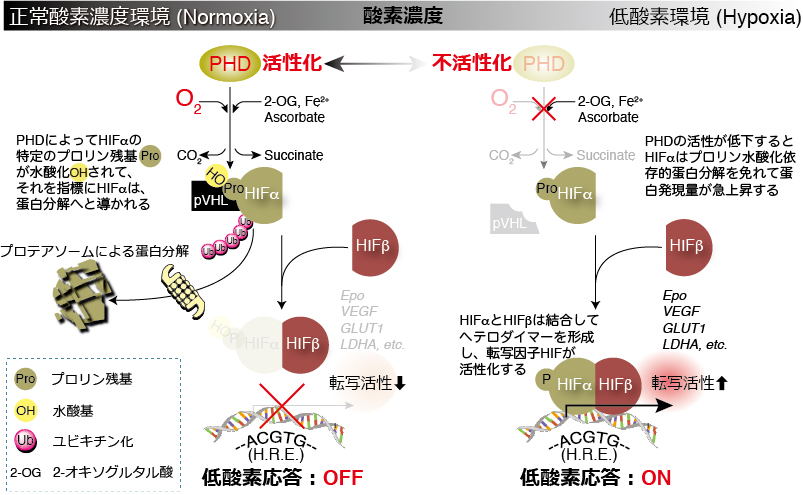
【図6】酸素が十分に利用出来る環境(normoxia)においてはPHDはプロリン水酸化酵素として機能しており、HIFαの特定のプロリン残基を水酸化することで、HIFαを蛋白分解へと導くため、HIFは転写因子としての活性が抑えられ、低酸素応答はOFFとなります。
一方で、低酸素環境 (hypoxia)においてはPHDの酵素活性が低下し、HIFαを分解に導くプロリン残基の水酸化が抑制され、ユビキチン化→プロテアソームにおける蛋白分解を回避したHIFαの発現量は細胞内で急速に上昇し、HIFβと結合してヘテロダイマーを形成し、転写因子HIFが活性化し、低酸素時に必要な遺伝子群の転写を活性化するため低酸素応答はONとなります。
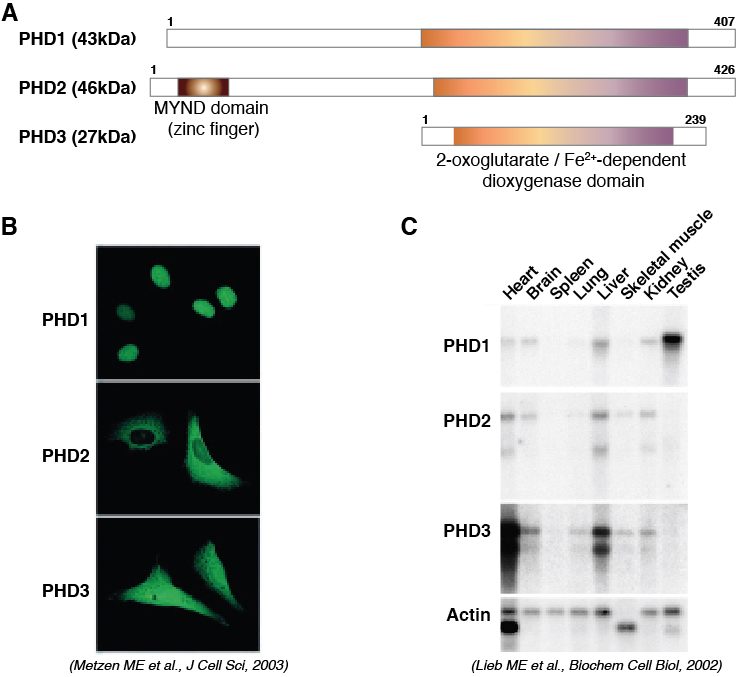
【図9】ですが、肝細胞において、この3つのPHDのうちPHD2を阻害(この場合、遺伝子を破壊しています)しただけでHIFを活性化させることが出来る(A)ので、in vivoにおいてはPHD2がドミナントな酵素であると言えるでしょう。因みに、PHD1やPHD3を欠損させたマウスがvaiableなのに対して、PHD2を欠損させたマウスは胎生致死でもありますので、PHD2は個体の発生に必須な遺伝子であることがわかります(Minamishima YA, et al., Blood 2008, Minamishima YA, et al., MCB 2009, Minamishima YA and Kaelin WG Jr, Science 2010)。
PHD2の欠損はHIFの発現を上昇させますが、そのHIFがまたPHD3を(細胞によってはPHD2も)誘導することや、PHD2とPHD3を破壊した肝細胞よりも、それに加えてPHD1も破壊した肝細胞のほうがHIF1αの蓄積が多い(図9Aの右側2レーンを参照)ことから、PHD1もin vivoでHIFを負に制御していることが解ります。
このように、生体内にはなんとかしてHIFの恒常的な活性化を抑制しようとするネガティヴ・フィードバック機構が存在することが解ります(Minamishima YA, et al., MCB 2009)。
それでは、何故、我々の身体はHIFの(あるいは低酸素応答の)恒常的な活性化を回避したがるのでしょうか?「生体内にはなんとかしてHIFの恒常的な活性化を抑制しようとするネガティヴ・フィードバック機構が存在する」のは、HIFの恒常的な活性化は生体にとって有害だからなのでは?と考えるのが普通でしょう。
以下に、慢性的な低酸素応答が引き起こす問題について解説します。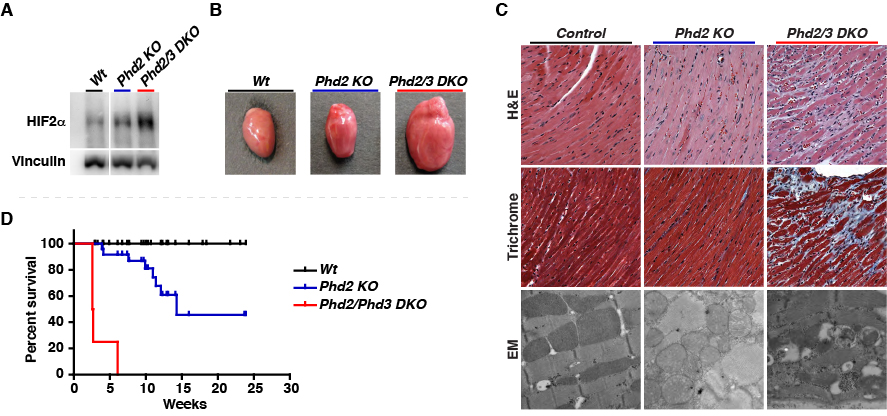
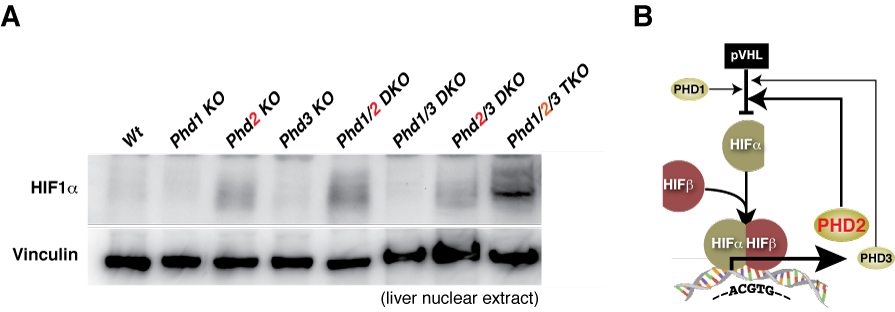
【図10】成体でPhd2やPhd3遺伝子を破壊したマウス(右)の心臓では、恒常的にHIFが活性化してしまっており(A)、著明な心肥大を来してしまいます(B)。心筋組織中では心筋細胞が脱落して高度に線維化し(上段のH&E染色と、中断のTrichrome染色)、下段の電顕所見(EM)ではミトコンドリアの損傷が著しく(C)、マウスは早期に死亡してしまうことがわかりました(D)。
Phd2 flox/flox;
Phd3 +/+;
CAG-Cre-ER(-)
Phd2 flox/flox;
Phd3 +/+;
CAG-Cre-ER(+)
Phd2 flox/flox;
Phd3 -/-;
CAG-Cre-ER(+)
【図11】低酸素応答が慢性的に活性化されたマウスの心エコー(長軸断層像)
(A) コントロール。(B) Phd2遺伝子をタモキシフェン投与によって破壊したマウス。軽度の心室腔の拡張が認められます。(C) Phd2に加えPhd3も欠損したPhd2/3ダブルノックアウトマウス。心室壁が薄く、心室が著明に拡張し、心収駆出率が低下し、拡張型心筋症に酷似した心不全に陥ってしまっていることがわかります。(C)のPhd2/3 DKOマウスでは、Phd2の欠損を代償するPhd3も欠損しているので、【図10A】のようにHIFαのプロリン水酸化が著しく低下してHIFが恒常的に活性化しているので、酸素センサー分子PHDの機能を慢性的に抑制し続けて低酸素応答を恒常的に活性化させると、拡張型心筋症に類似した心不全が原因で個体が早期に死亡してしまうことが明らかとなりました (Minamishima YA, et al., Blood 2008, Minamishima YA, et al., MCB 2009)。
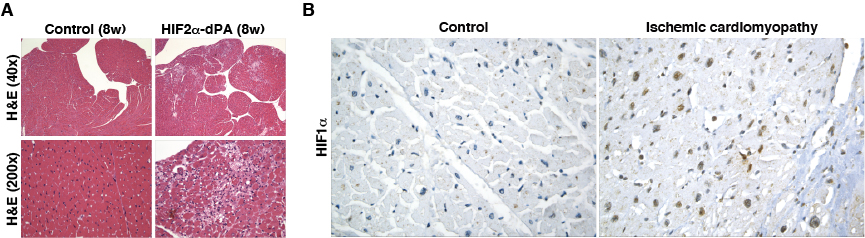
【図12】PHDによる水酸化部位のプロリンをアラニンに置換したHIF2α (HIF2α-dPA)を心筋特異的に発現させたマウスの心臓では、ユビキチン化→蛋白分解を回避出来る蛋白分解耐性のHIF2αが恒常的に発現しており、その結果、心筋細胞が激しく脱落し、心組織が高度に線維化してしまっていることがわかります。すなわち、心筋の線維化は慢性的なHIFの活性化で引き起こされることが証明されたことになります(A)。
また、過去に心疾患に罹患したことのない患者(Control)と、過去に心筋梗塞を経験した虚血性心筋症(Ischemic cardiomyopathy)の患者の、剖検心筋組織におけるHIF1αの免疫染色。Ischemic cardiomyopathy患者の心筋細胞ではHIF1αが顕著に蓄積しており(B)、心筋梗塞後に問題となるischemic cardiomyopathyとは低酸素応答の慢性的な活性化によって引き起こされる可能性を示しました(Moslehi J*, Minamishima YA*, et al., Circulation 2010)。
ですが、基本的に低酸素応答は低酸素環境から身を守るための生体防御メカニズムであったはず。上手に使えばなにかの病気の治療に応用出来ないだろうかと考えました。
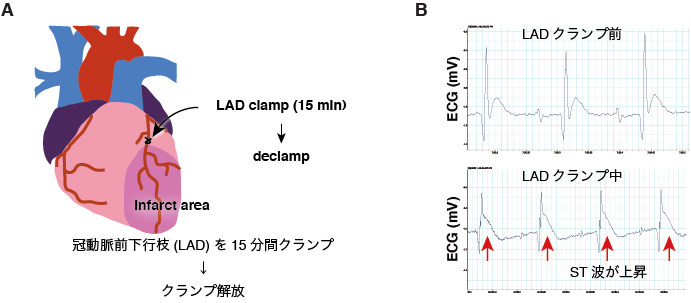
【図13】マウスの心臓の左冠状動脈前下行枝(LAD; left anterior descending artery)を15分間遮断(clamp)して血流を遮断したのちにdeclampして血流を再開させることによって、心筋梗塞モデルを作製することが出来ます(A)。Clampしている間は、心電図でST波が上昇しており、心臓が虚血に陥っていることが解ります(B)。
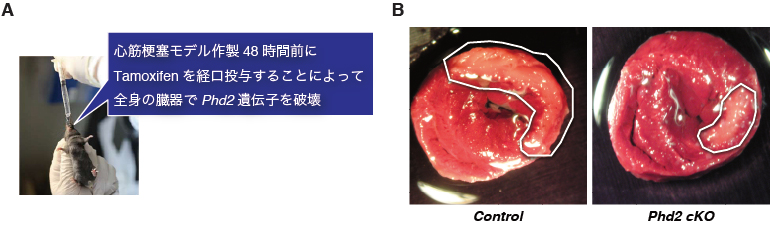
【図14】このマウスの心筋梗塞モデルを、48時間前にタモキシフェン投与によって全身の臓器でPhd2遺伝子を破壊したマウスで作製してみました(A)。その結果、Phd2遺伝子を破壊したマウスでは、コントロールと比較して心筋梗塞巣のサイズが有意に縮小しており、また、心機能も保たれていました。このことから、心筋梗塞のような虚血再灌流障害が問題となる状況に於いては、Phd2を阻害して低酸素応答を活性化させると生体にとって有利に働くであろうことがわかりました。
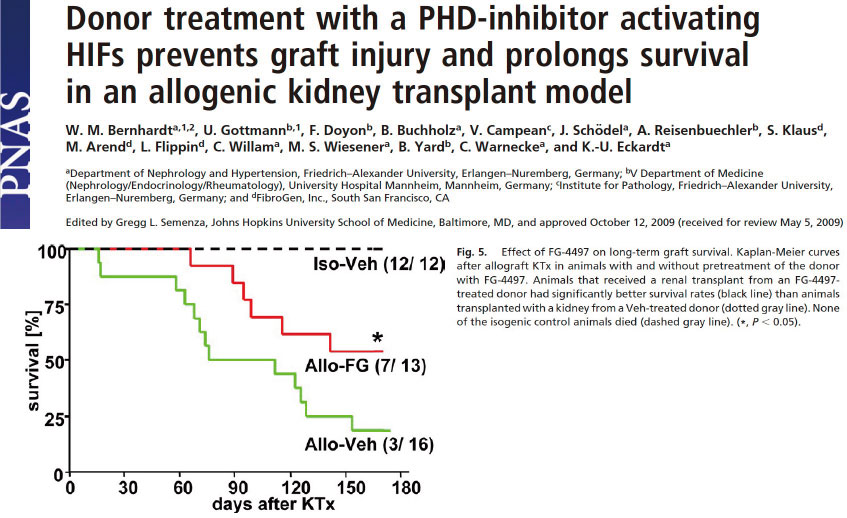
【図15】同様に、虚血再灌流障害が問題となる疾患・病態といえば、臓器移植が筆頭にあげられるでしょう。ラットの腎移植アログラフトモデルにおいて、ドナーにPHD阻害剤(FG-4497)を投与すると、移植グラフト生着率・個体の生存率がVehicle投与群と比較して有意に高いとの報告があります(Eckardt KU, PNAS 2000)。このように、低酸素応答を活性化すると、移植臓器の保護・生着率/ドナーの生存率の改善が期待出来ることが解ります。
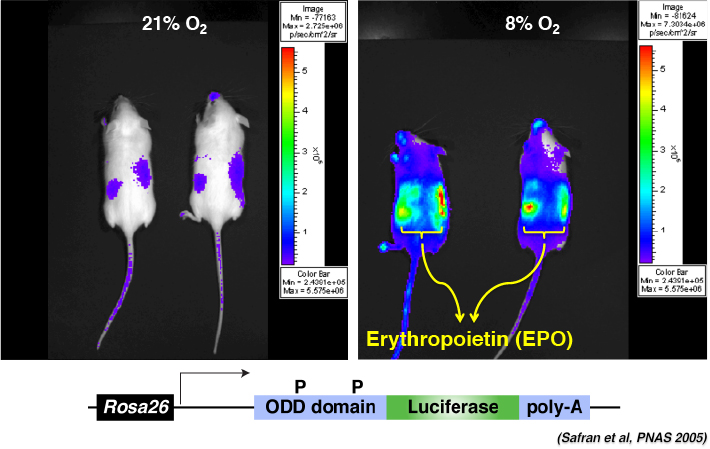
【図16】低酸素状態に陥った細胞のみがLuciferaseが発現するマウスを正常酸素濃度(左。21% O2)から低酸素環境(右。8% O2)へ移すと、腎臓に一致した部位に強いシグナルが検出される(=低酸素状態になっている)ことが解ります。すなわち、腎臓という臓器は、低酸素に対して非常に敏感な臓器であることが解ります。このような「低酸素に敏感な臓器に赤血球を増やす造血ホルモンであるエリスロポエチン(EPO)を産生させる」という我々の身体の低酸素応答プログラムは、非常に理に適っていると言うことができるでしょう。
逆に言えば、慢性腎臓病(CKD; chronic kidney disease)などで腎機能が低下してしまうと、腎におけるEPO産生が低下し、患者さんは腎性貧血に苦しむことになります。腎性貧血に対しては数週間に一度リコンビナントEPOを投与すれば良いのですが、未だに内服薬がないので患者さんは病院へ注射を打ちに行かなくてはならないこと、うっ血性心不全、高血圧、不整脈などの副作用が知られていること、それに加えて高い医療費などが問題となっていました。
そこで我々は、EPOが胎生期は肝細胞によって産生されていることに着目しました。たとえ腎機能が低下してしまっても、肝細胞にもう一度EPOを産生して貰えば、腎性貧血の治療になるかも知れない、と考えた訳です。
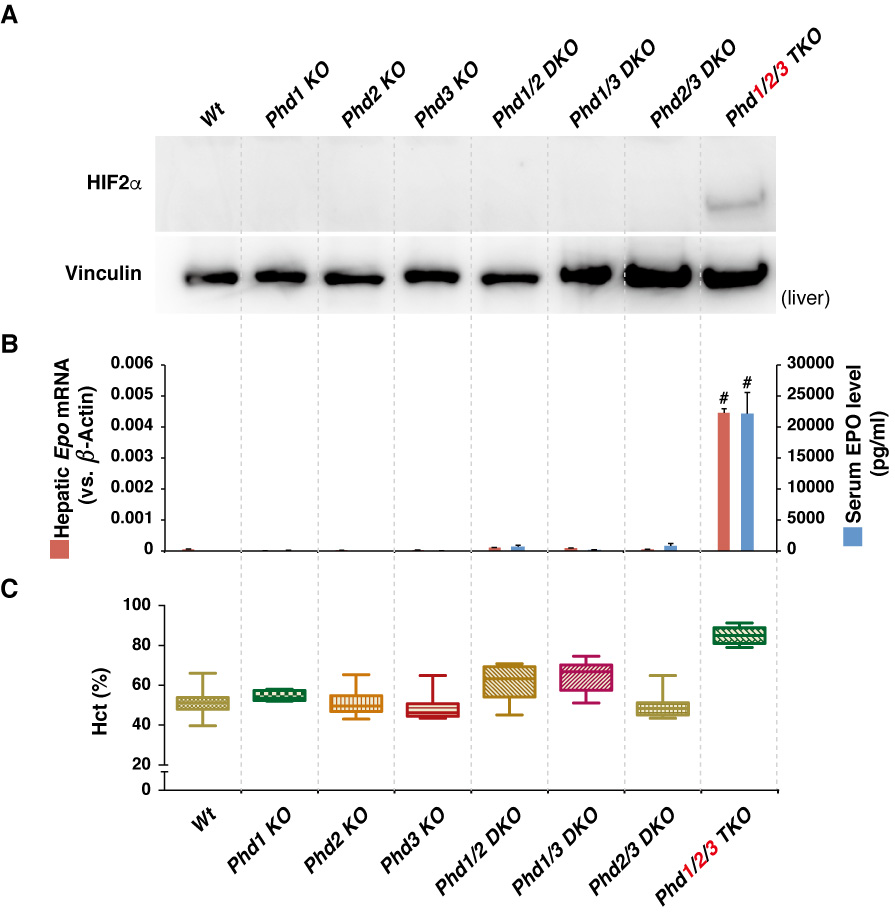
【図17】肝臓においてPHD1〜3を破壊したマウス(Phd1/2/3 TKO)においては、EPOを誘導するHIF2αの発現量が肝臓で著明に上昇し(A)、肝臓におけるEpoのmRNAの転写量も増加し(B. 赤のグラフ)、血中のEPO濃度も増加し(B. 青のグラフ)、赤血球が増加してヘマトクリット値(Hct)が顕著に上昇すること(C)がわかりました。すなわち、PHD1, PHD2, PHD3の3つの酵素を阻害するだけで、成体の肝細胞が再びEPOを産生し赤血球数が増加することが証明された訳です(Minamishima YA and Kaelin WG Jr, Science 2010)。
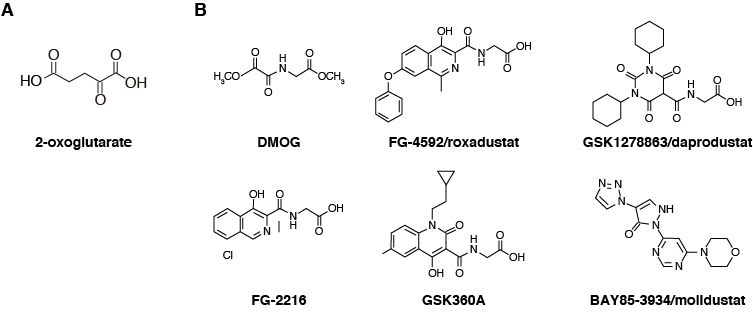
【図18】このPHDを阻害する薬剤が様々な会社から開発され、EPO補充療法に替わる「腎性貧血」の治療薬として臨床治験中でしたが、2019年9月にFG-4592/Roxadustatが(透析導入中の)腎性貧血の治療薬として本邦でも認可されました。低酸素応答の分子メカニズムを、ヒトの病気の治療へと応用した最初のケースになります。基本的にはどれも2-オキソグルタル酸(上段)のアナログなので、どの薬剤も2-オキソグルタル酸に構造が類似していることが解ります(molidustatを除く)。
PHDを阻害すればHIFが活性化されて低酸素応答をONにすることが出来る、ということは、酸素が十分あるnormoxiaの状態においても、細胞や個体に「いま自分たちは低酸素環境に晒されているのだ」だと勘違いさせて低酸素応答をONにさせることが出来る、ということです。このような薬剤を用いれば、低酸素応答をin vivoで自在にON/OFF出来るようになります。
我々の研究室では、このアイデアを応用して、低酸素応答を活性化させれることが様々な疾患の治療法へと応用出来るのではないか?と考え、低酸素応答を標的とした各種疾患治療法の開発までを視野に入れた基礎研究を展開していきます。
例えば、敗血症や重症感染症などに合併する乳酸アシドーシスに対して、PHD阻害剤を投与することで、肝臓や腎臓における乳酸からの糖新生が活性化し、乳酸アシドーシスの生存率を劇的に改善させることについて発表してきました(Suhara T et al., PNAS 2015; Oyaizu-Toramaru T e al., MCB 2017)。詳しくはこちらの総説をどうぞ。
また、冒頭にもご紹介したとおり、この低酸素の生物学は非常にホットな研究分野です。有り難いことに、Nobel賞受賞講演でも南嶋の研究を紹介して頂いてます。
- 硫黄代謝物による生体内代謝制御機構の解明
酸化還元、呼吸・エネルギー代謝などに対して強力な生理活性を持つ含硫代謝産物について、その固体での役割や存在意義について考察していきます(東北大学・Massachusetts General Hospital, Harvard Medical School・生理学研究所・慶應義塾大学との共同研究)。
- 生理活性脂質の産生/分解機構や生体内での機能の解明
細胞が刺激を受けると、膜のリン脂質が分解されて種々の生理活性脂質が作られます。その作用の大部分は細胞膜に存在するGタンパク質共役型受容体 (GPCR)を介するもので、神経、呼吸、循環、生殖などほとんどすべての生命活動に関与しています。
我々のラボでは、生理活性を持ったリン脂質の生合成や生理的機能を解明するための生化学的研究を行っていますが、主に
- スフィンゴシン1-リン酸 (S1P; Sphingosine 1-phosphate)
- リゾホスファチジン酸 (LPA; lysophosphatidic acid)
- 環状ホスファチジン酸 (cPA; cyclic phosphatidic acid)
に着目して研究を進めています。
研究手法は酵素学、生化学、機器分析学(質量分析を含む)、分子生物学、細胞生物学と多岐にわたります。生理活性脂質は炎症・アレルギー反応・免疫疾患・神経疾患・腫瘍などの病態形成に深く関与しているため、病態の解明と新しい治療法の開発を視野に入れた研究を心がけています(大日方・和泉・大嶋・南嶋)。
- 代謝による細胞老化の制御機構の解明
「すべての生物は細胞で構成されており、生物の構造的・機能的基本単位は細胞である」とは1800年代前半に既に提唱されていた、所謂「細胞説」です。我々の身体が健康であるためには、身体を構成する細胞の自己複製・増殖・分化が厳密に制御されていなくてはなりません。遺伝情報が保存されているゲノムDNAの損傷は、細胞の老化やがん化だけでなく様々な疾患の発症へと直結するため、DNA傷害の検知機構と損傷を受けたDNAの修復機能を理解することは、がんの予防や治療を考える上で非常に重要です。当研究室では、ゲノムDNA末端のテロメアの保護機構に着目し、分子生物学・遺伝学・生化学・分析化学的手法を駆使してDNA修復の分子メカニズム解明を目指しています(小西)。
また、正常な細胞というものは無限に増殖出来るものではありません。ヒトの様々な臓器から分離された細胞は、細胞分裂に伴うテロメアの短縮などの理由で、その細胞に固有な分裂回数に達したら増殖を停止してしまいます(ヘイフリック限界: Hayflick limit)。それ以外にも、各種ストレス(DNA傷害、酸化ストレスなど)によって、細胞は増殖を停止しますが、その現象は細胞老化と呼ばれています。
しかし、細胞の老化は癌化を防ぐためのセーフティ機構なのか、それとも老化した細胞が癌化するのか。正常な細胞、老化した細胞、癌化した細胞は何が違うのか。そもそも細胞の老化の定義とは細胞の非可逆的な増殖停止のことなのか?ひとたび老化した細胞はもう二度と増殖しないのか?...などなど、実はまだよく解らないことだらけなのです。
当研究室では、正常な細胞と老化した細胞とのエネルギー代謝の違いなどを較することによって、「いったい細胞の老化とは何なのか?」というその素朴な疑問に対する答えを追求していきます(小西・入江・南嶋)。
- 癌細胞特異的な代謝機構の同定と治療標的の探索
正常細胞・がん化した細胞のメタボローム解析から、がん化した細胞に特異的な代謝産物や代謝経路を既にいくつか同定しています。がん特異的な代謝システムの全貌解明と、その癌特異的代謝メカニズムを標的とした次世代癌治療法の開発までを視野に入れた研究を展開しています(小西・南嶋)。
- 質量分析計を用いたプロテオーム解析・メタボローム解析(リピドーム解析も含む)による細胞内生理的/病理的イベントの分子メカニズム解明
遺伝子発現量(mRNAの定量)だけを評価しても、実際に細胞内で何が起きているのかを正確に理解することは出来ません。シグナル伝達や遺伝子発現の変化の結果、様々なタンパク質の存在量やリン酸化などの修飾、代謝産物(糖、アミノ酸、脂質、核酸、などなど...)の増減が、細胞の機能を制御しているからです。蛋白や代謝産物などを定量することが出来るこの質量分析技術は、医学・生命科学研究で必須の技術の1つですが、当研究室では最新の質量分析器を駆使したプロテオーム解析・メタボローム解析(リピドーム解析も含む)をメインの研究手法の1つに据えた医学・生命科学研究を展開しています(大日方・小西・大嶋・南嶋)。
興味を持たれた方、どうぞお気軽にご連絡ください。 ![]()


